T-BAS v2.2 User Manual
Table of Contents
- Color Editor
- Data Standardization
- DeCIFR REST Server and API
- De novo single or multi-locus phylogenetic analysis
- References
- Appendix
- Description of Terms
1. Color editor
The purpose of the color editor is to allow the user to select preferred colors for the layout of the tree. When T-BAS creates a tree it randomly assigns colors to attributes from all colors in the spectrum. For each attribute, the rows in the legend are arranged by color so that the user can find the label of a color by looking in the legend. The colors can be changed in the color editor. However, the order of entries in the legend remains as for the original colors assigned. There is no limit to how many values or attributes can be edited.
To change the colors, click the color editor button and the color editor window will pop up.
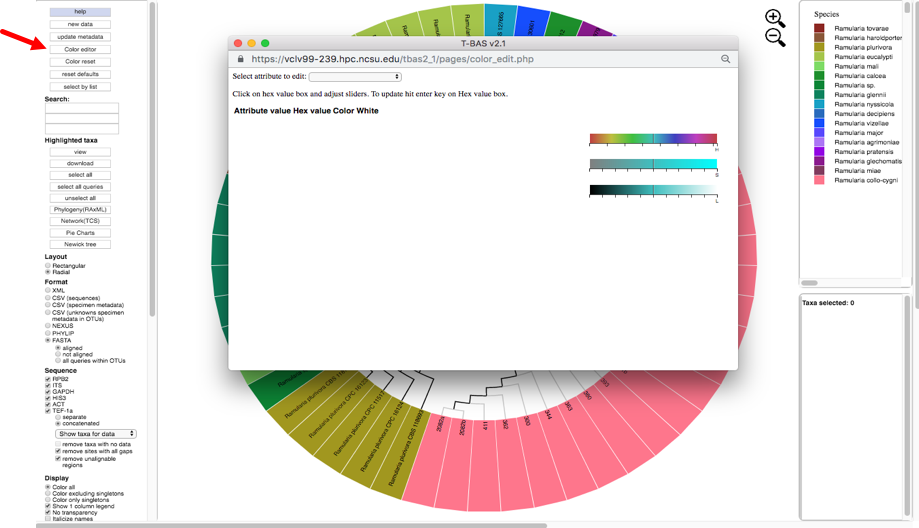
There are two ways to change the colors. Colors can be selected on the color bars or inputting aknown hex color value.
To change the color using the HSL (hue, saturation, lightness) color bars, slide the center vertical black line (while holding down the left mouse button) on one of the 3 bars to the left or right. One or all three bars can be modified in order to display the desired color. The letter under the corresponding bar indicate the following: H (hue), S (saturation), L (lightness).
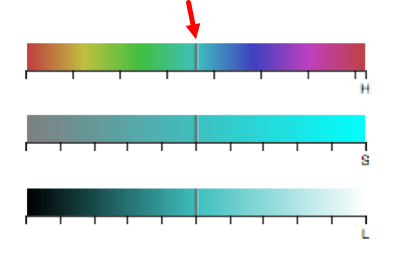
Selecting a specific attribute in the pull-down menu will display the current color arrangement on the tree. Here the hex values can be changed, if known. Hex values can be searched online or can be viewed here. Enter the value into the box and press Enter/Return.
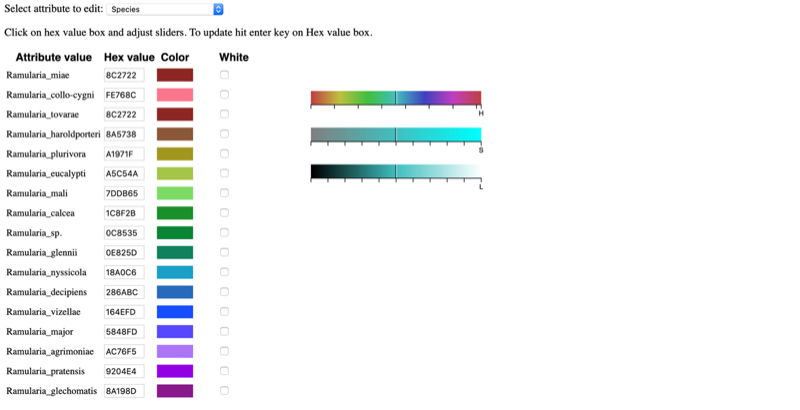
If the value is not known, click inside a box of an attribute to be changed, and select a new color on the color bar or adjust the vertical black lines until a desired color appears. For the change to take effect, the cursor must be inside the box that has the edited color value and press Enter/Return. The colors will then be updated in the color editor, in the tree, and in the legend. To select the color white, click the box in the last column.
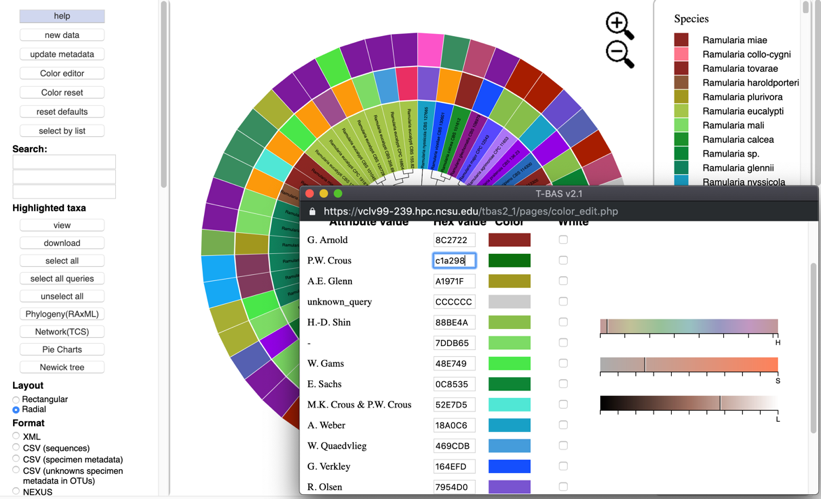
Clicking the color reset button will undo all changes.
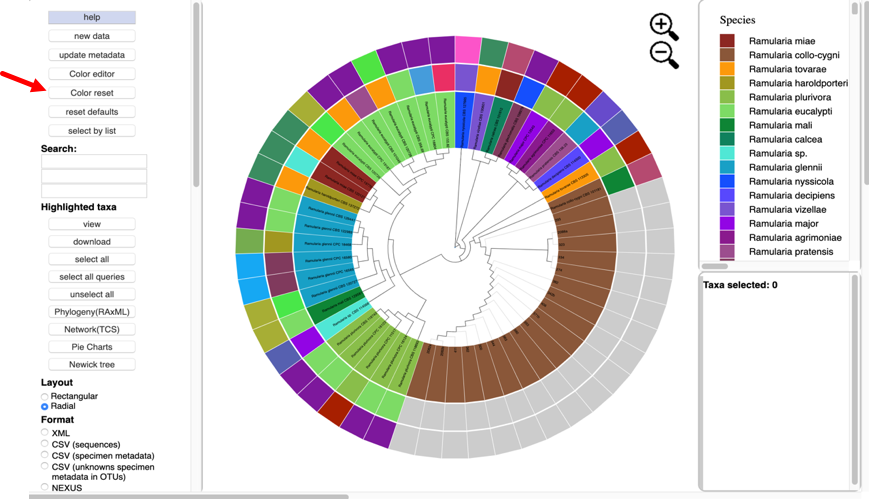
To copy a color scheme from one tree to another, copy hex values and then enter them manually in the color editor on the next tree.
2. Data Standardization
In T-BAS, DNA sequences and associated specimen metadata are phylogenetically placed on curated multilocus reference trees and the placement results are saved as Metadata Enhanced PhyloXML (MEP) files. The MEP format allows placements and associated specimen attributes (e.g. host, locality, environmental traits) to be readily viewed, archived and importantly analyzed within a phylogenetic context. MEP files are structured to adhere to the minimum information about any (x) sequence (MIxS) family of standards defined by the Genomic Standards Consortium. A template is provided for users to fill in and submit when performing a phylogeny-based placement in T-BAS. Additional categories of metadata information can be added. MIxS headers and metadata are saved in MEP files as defined in the XML schemas below. The use of MEP files ensures interoperability and retrieval of relevant sequences and metadata for downstream applications. This standardization provides a consistent handling of the data and is currently used by T-BAS and other tools in the DeCIFR toolkit. MEP is based on XML (Han & Zmasek 2009), a widely used markup language for representing and sharing information, and PhyloXML, an extension of XML with custom tags for describing evolutionary trees or networks.
The standard pre-defined XML schema for phyloXML is used as a starting point for validating MEP files. PhyloXML includes a phylogeny element that saves the tree information and associated alignments. MEP extends this by adding (1) an OTUs tag that saves the taxonomic assignments, associated query metadata and sequences for each OTU, (2) a tag to each clade that is a leaf in the tree and saving the metadata for that leaf, and (3) a gene tag that saves the locus name, the number of sequence characters, and the positions of the excluded unaligned character set (i.e. exset) for each alignment
MEP uses two associated schema definitions:
- cifr_phyloxml.xsd to show how custom tags are added to PhyloXML.
- cifr.xsd to define custom tags in the http://www.cifr.ncsu.edu namespace.
The MEP schema includes new tags: cifr:otus, cifr:attributes, and cifr:genes.
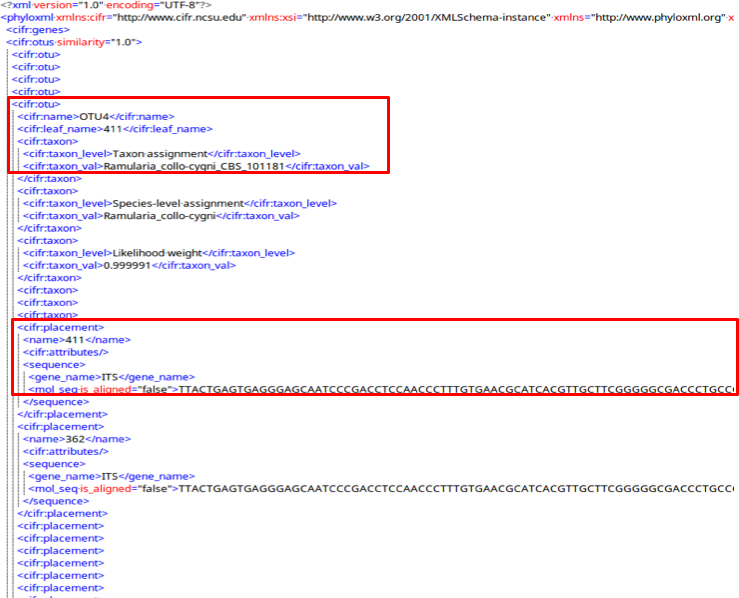
cifr:otus A cifr:otu tag saves all the information in the OTUs of the submitted samples.
A cifr:otu tag contains a cifr:name, cifr:leaf_name, and a cifr:taxon tag.
The cifr_taxon tag contains cifr:taxon_level and cifr:taxon_val tags with placement information for this OTU.
Also in the cifr:otu are cifr:placement tags with attributes and unaligned sequences for each sample in the OTU.
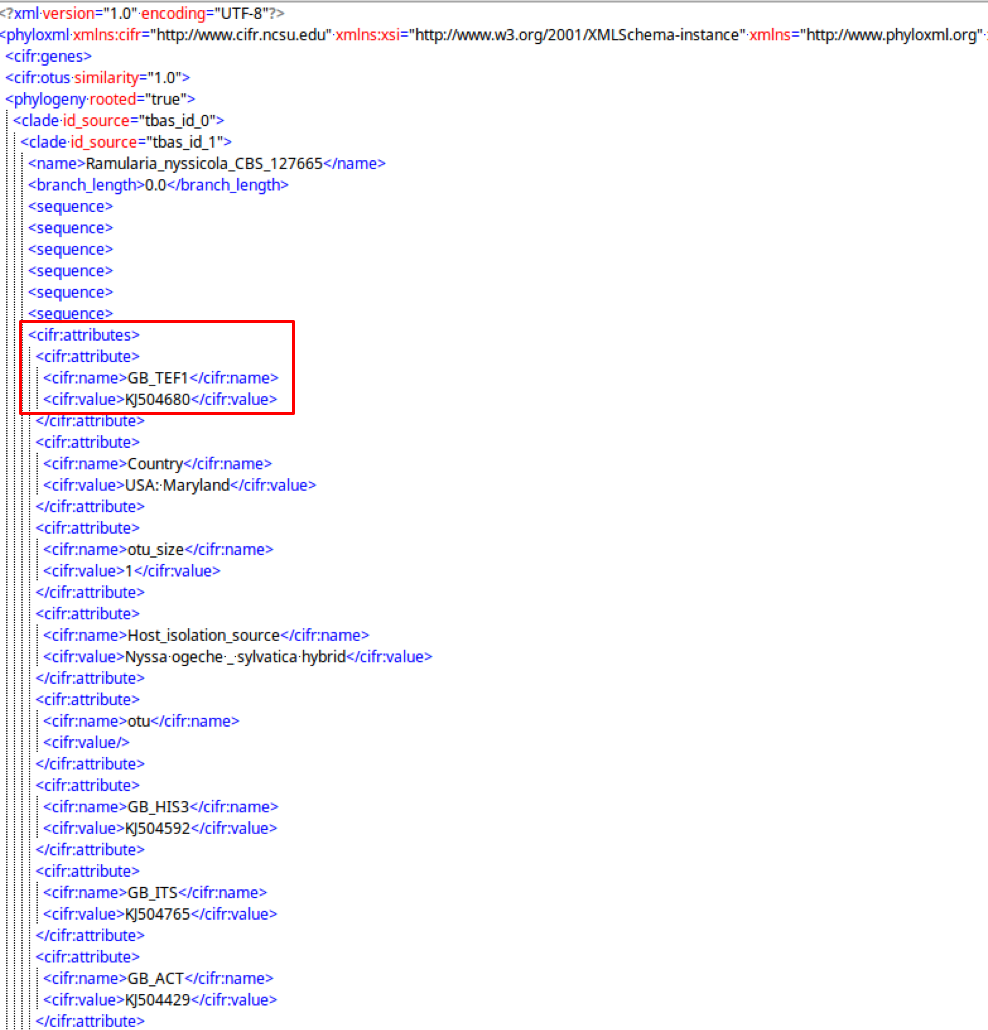
cifr:attributes
A cifr:attributes tag contains information for specimen metadata in the tree structure.
The cifr:attributes tag contains cifr:attribute, which contains cifr:name and cifr:value.
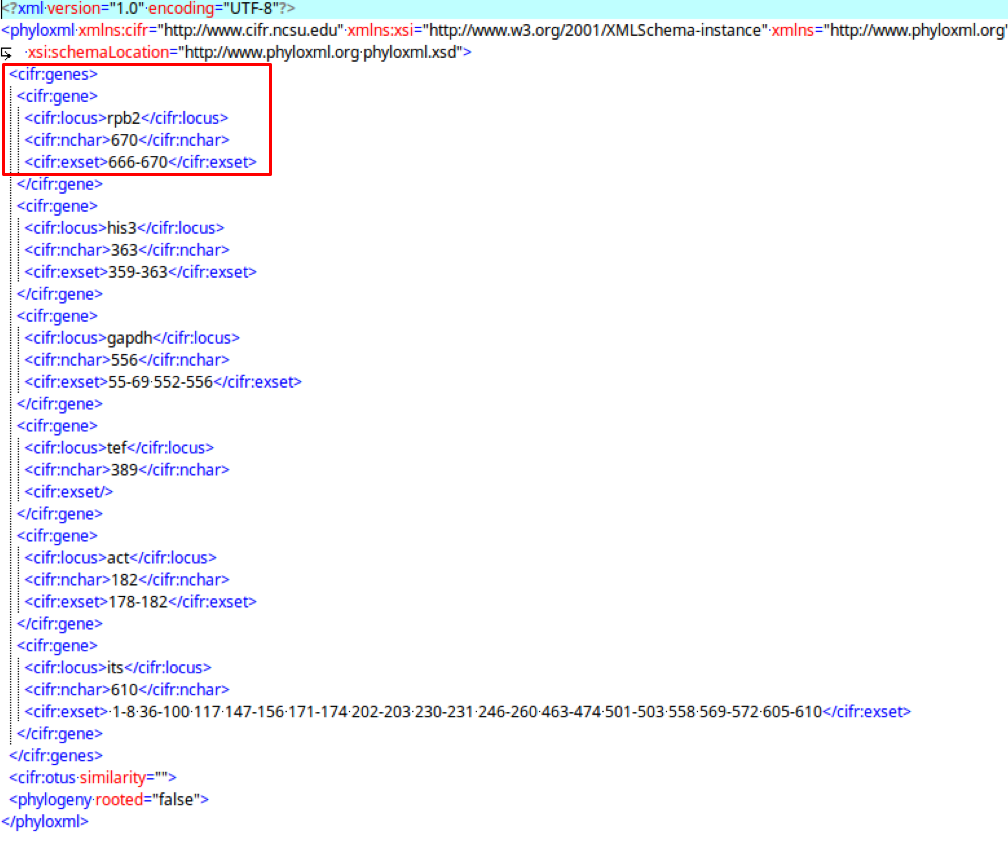
cifr:genes
The cifr:gene tags saves metadata of the alignments.
The cifr:genes tag contains cifr:gene, which contains cifr:locus, cifr:nchar, and cifr:exset.
3. DeCIFR REST Server and API
REST Server:
The Representational State Transfer Application Program Interface (REST) server can be used to retrieve tree information from Metadata Enhanced PhyloXML (MEP) files. This was released on github (https://github.com/ncsu-decifr/decifr-rest) using an open source BSD 3-Clause License. The DeCIFR REST server allows a user to share information about placements from T-BAS with other users via the web. The program is written to run in a Python 3 virtual environment and uses Flask (https://palletsprojects.com/p/flask/). Installation instructions are included.
REST API:
The DeCIFR REST API service allows access to results of a previous run of T-BAS v2.1.1 via HTTP with a browser request, programmatically, or using a command line tool such as CURL. More information is available at https://tools.decifr.hpc.ncsu.edu/rest.
Docker users only:
Opening the URL to /list returns a list of run IDs of all the XMLs in the folder.
Clicking on a Run ID link (ie; 3F7THARX) will allow the leaves, queries, and OTUs to be viewed without opening the tree.
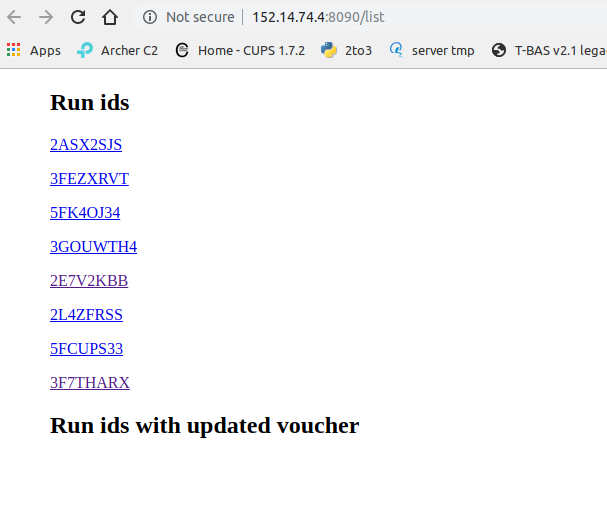
Click on ‘leaves’ to see the sample names that are present in a tree.

Click on the metadata link for Ramulaira_calcea_CBS_101612 (#2).

The metadata for that sample will be shown.
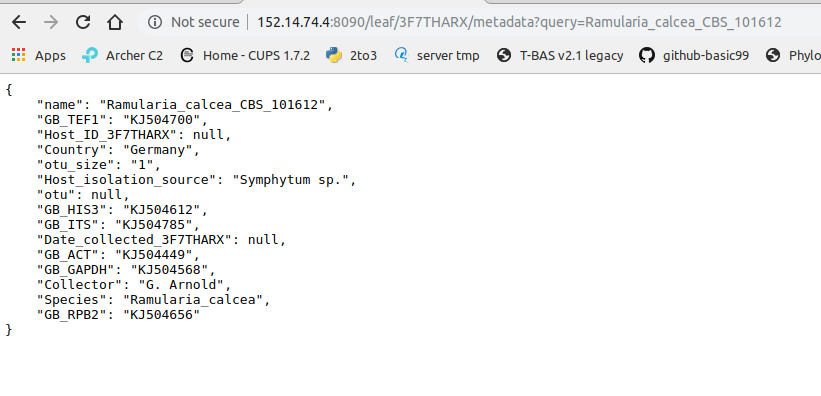
Clicking on “queries” will display the query and the tree placement information:
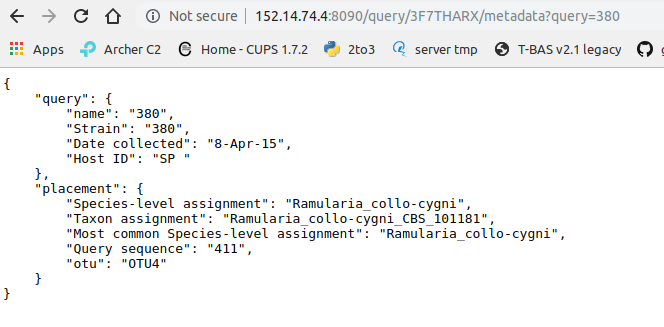
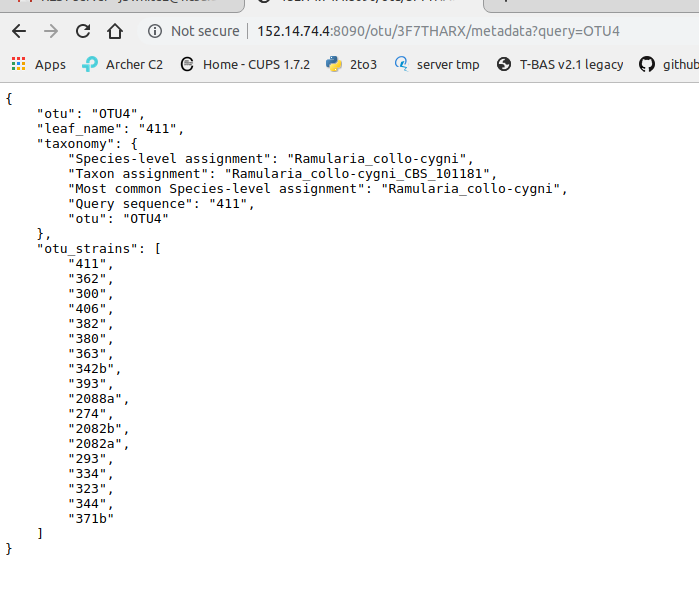
Clicking on OTU will display the following information:
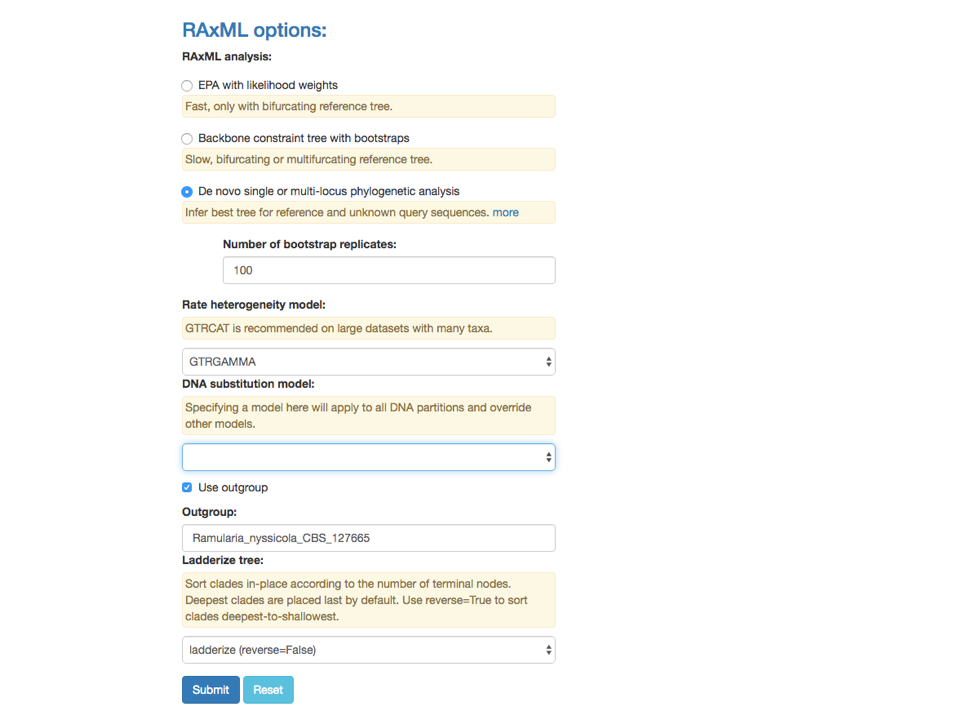
4. De novo single or multi-locus phylogenetic analysis
This feature under the RAxML options can be used to Infer best tree for reference and unknown query sequences. Potential applications include: (1) inferring trees for species delimitation using the Genealogical Concordance Phylogenetic Species Recognition (GCPSR) concept (Taylor et al 2000), and (2) inferring an input tree for Poisson Tree Processes (PTP) model to delimit putative species (Zhang et al 2013).
5. References
Section 1 Color Editor
https://www.compuhelpts.com/Color_Codes_1.pdf
Section 2 Data Standardization
Han MV, Zmasek CM (2009) phyloXML: XML for evolutionary biology and comparative genomics. BMC bioinformatics 10, 356.
Section 4 De novo single or multi-locus phylogenetic analysis
Taylor, J.W., D.J. Jacobson, S. Kroken, T. Kasuga, D.M. Geiser, D.S. Hibbett, et al. 2000. Phylogenetic species recognition and species concepts in fungi. Fungal Genet Biol 31: 21-32. doi:10.1006/fgbi.2000.1228.
Zhang, J., P. Kapli, P. Pavlidis and A. Stamatakis. 2013. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29: 2869-2876. doi:10.1093/bioinformatics/btt499.
6. Appendix
Description of Terms
| Term | Description |
|---|---|
| Backbone constraint tree with bootstraps | RaxML method |
| Bifurcating tree | Tree where each node has 2 children |
| BLAST | Basic Local Alignment Search Tool, used to match unknown sequences to known sequences in database |
| De novo phylogenetic analysis | RaxML method |
| EPA with likelihood weights | RaxML method that places sequence on edges of existing tree |
| FASTA | A file sequence format for unaligned data |
| Genetic distance cutoff | Value used by custom algorithm to exclude divergent species from placement |
| GTRCAT (Rate heterogeneity model) | Faster model than GTRGAMMA that uses a different approximation to capture rate heterogeneity across sites |
| GTRGAMMA (Rate heterogeneity model) | General Time Reversible (GTR) model with Gamma distributed rates across sites |
| ITS | Internal transcribed spacer locus |
| Labels: Display Names | Node-click context menu, display leaf names in selected clade in large trees. Tree with greater than 2000 leaves do not display names for performance reasons. |
| Labels: Likelihood Weight | Node-click context menu, click on leaf of EPA placement will show all leaves attached to the edge that gives 95% cumulative weight. |
| Ladderize tree | Sort tree leaves from deepest to shallowest or reverse |
| Locus (Loci) | A location on a chromosome |
| LSU | Large subunit locus |
| MEP | Metadata Enhanced PhyloXML format that is a valid phyloXML with added tags for use in T-BAS and DeCIFR |
| Metadata: Download | Node-click context menu, download data of selected according to selections in format and sequence |
| Metadata: View | Node-click context menu, view data of selected according to selections in format and sequence in pop-up window |
| Multifurcating tree | Tree where each node can have multiple children |
| NEWICK | A standard for representing trees |
| NEXUS | A file format with multiple uses, can contain trees and alignments |
| OTUs | A grouping of sequences into percent similarity |
| Outgroup | Leaves of a tree placed in a distinct clade, used to root tree |
| PHYLIP | A file format for aligned sequence data |
| PhyloXML | XML language designed to describe phylogenetic trees (or networks) and associated data |
| Query sequences | Unaligned unknown sequence data |
| Rate heterogeneity model | A phylogenetic model that accounts for evolutionary rate heterogeneity |
| RAxML | Software tool used to place alignment on a tree, plus some other utilities |
| Reference set | A set of tree, alignments, and metadata of known species at a specific taxonomic level used for placement |
| Taxa: Select All | Node-click context menu, select all leaves on tree |
| Taxa: Select(unselect) | Node-click context menu, select or unselect all leafs in clade |
| Taxa: Unselect All | Node-click context menu, unselect all leaves on tree |
| Tree: Collapse(expand) | Node-click context menu, collapse clade into a single node. Collapsed clade appears as a small circle. Click on this circle to restore clade. |
| Tree: Network (TCS) | Node-click context menu, create TCS network of all query strains in clade |
| Tree: Newick tree | Node-click context menu, download newick tree of selected clade in either phylip or NEXUS format |
| Tree: Phylogeny (RaxML) | Node-click context menu, create de novo tree of selected clade |
| Tree: Pie Charts | Node-click context menu, create pie charts to show relationships of selected attributes |
| Tree: Subtree (new window) | Node-click context menu, view subtree of selected clade in new window |
| Tree: Subtree(tree) | Node-click context menu, view subtree of selected clade |
| UNITE | Database of fungal ITS for BLAST |